-
11e
CHAPTERRenal Tubular Acidosis
Surjit Tarafdar
ABSTRACT
Renal tubular acidosis (RTA) can be described as a group of disorders characterized by normal anion gap (hyperchloremic) metabolic acidosis with either hypokalemia or hyperkalemia. Both proximal RTA (type 2 RTA) and distal RTA (type 1 RTA) are associated with hypokalemia and result from tubular inability to reabsorb bicarbonate or excrete hydrogen, respectively. With the similar biochemical presentation, the only definitive way to distinguish between the two conditions is the urine pH as the urinary pH is >5.5 in type 1 RTA while it is <5.5 in type 2 RTA. While type 1 RTA is quite common in Sjögren’s syndrome, one must always consider myeloma in the differentials of type 2 RTA.
Type 4 RTA is associated with hyperkalemia and is often due to diminished production or reduced tubular effectiveness to aldosterone and is commonly associated with diabetes mellitus as well as drugs like angiotensin-converting enzyme (ACE) inhibitor, angiotensin receptor blocker (ARB), or aldosterone antagonists.
INTRODUCTION
Renal tubular acidosis (RTA) refers to a group of disorders characterized by normal anion gap (hyperchloremic) metabolic acidosis with either hypokalemia or hyperkalemia and a relatively well preserved glomerular filtration rate (GFR).
• Normal anion gap metabolic acidosis with hypokalemia:
○ Proximal RTA (type 2 RTA) is caused by reduced tubular ability to reabsorb bicarbonate (HCO3–).
○ Distal RTA (type 1 RTA) is caused by defect in distal hydrogen (H+) excretion.
• Normal anion gap metabolic acidosis with hyperkalemia:
○ Type 4 RTA is due to either aldosterone deficiency or tubular resistance to the action of aldosterone. Less commonly this condition is caused by inherited or acquired defects in sodium (Na+) transport in the principal cells in collecting duct.
○ While type 4 RTA is the most common type of RTA, type 1 RTA is more common than type 2 RTA.
KIDNEY’S ROLE IN MAINTAINING ACID–BASE AND POTASSIUM BALANCE
The normal acid–base balance of the body depends on:
• Lungs: Alveolar ventilation removes carbon dioxide
• Kidneys: The tubules reclaim filtered HCO3– and excrete H+
The kidneys in a healthy adult filter about 4,200 mmol of HCO3– every day and all of it is reabsorbed in the renal tubules. While 85–90% of this HCO3– is reabsorbed in the proximal convoluted tubules (PCTs), the remaining 10–15% is reabsorbed in the distal nephron.
Action of Carbonic Anhydrase in Reclaiming HCO3– in the Proximal Convoluted Tubule (Fig. 1)
The renal tubular cells similar to every cell in the body are deficient in Na+ due to the basolateral Na+-K+-ATPase extruding 3 Na+ from the cell and pulling in 2 K+ (Fig. 2). At the level of the glomeruli, with a GFR of 125 mL/min, healthy adults filter about 180 L of water over a day. More than 90% of the filtered water is reabsorbed in the tubules because water follows Na+, with the tubular cells being hungry for Na+ due to the intracellular deficiency.
Sodium bicarbonate (NaHCO3) is freely filtered across the glomerular capillaries and dissociates in the lumen of PCT into Na+ and HCO3–; this Na+ is reabsorbed by the tubular cells in exchange for H+. In the tubular lumen, HCO3– combines with the H+ to form carbonic acid (H2CO3) under the influence of tubular carbonic anhydrase (CA). H2CO3 then splits into CO2 and water. Water is lost in the urine while the CO2 diffuses into the PCT cells and regenerates H2CO3 intracellularly after combining with H2O. Aided by the intracellular CA, this H2CO3 then splits to form H+ and HCO3–. This H+ is secreted into the PCT lumen in exchange for Na+ while the HCO3– diffuses into the peritubular capillaries. The net result is that both Na+ and HCO3– (from the filtered NaHCO3) are reabsorbed in the PCT cells. As the Na+ is reabsorbed, it drags along with it glucose, phosphate, uric acid, and amino acids.
An interference with the above would lead to bicarbonate loss in the urine causing type 2 RTA, as well as Na+ and the associated glucose, amino acids, phosphate, and uric acid loss in the urine, leading to glycosuria and proteinuria along with low plasma phosphate and uric acid (Fanconi syndrome).
An average healthy adult generates 50–70 mEq of acid per day from diet, most of which is excreted in the urine by tubular secretion. Excretion of this acid load requires urinary buffers to bind H+ as otherwise the urine would become supersaturated with excessive H+ and the pH would drop severely, inhibiting any more H+ secretion. Urine will not accept any more H+ from the tubules if its pH falls below 4.5.
The chief urinary buffers are ammonia-ammonium (NH3 and NH4+) and phosphate (HPO4-2 and H2PO4-1).
TYPES 1 AND 2 RTA
Type 1 RTA is caused by inability of the distal tubules to secrete H+. The kidney characteristically cannot lower the urinary pH to <5.5 due to deficiency of H+ in the urine.
Na+ is reabsorbed in the tubules along with an anion, such as chloride (Cl–) or bicarbonate (HCO3–), or in exchange for a cation, such as K+ or H+. As H+ secretion is impaired in type 1 RTA, the K+ secretion increases as a compensation leading to the hypokalemia.
While type 1 RTA may be inherited, acquired causes are more common and include Sjögren’s syndrome, systemic lupus erythematosus (SLE), hypergammaglobulinemic states, primary biliary cirrhosis, autoimmune hepatitis, hypercalciuria, obstructive uropathy, and renal transplantation (Table 1). Drugs like amphotericin can lead to type 1 RTA by causing excessive back leak of H+ in the distal tubules.
Sjögren’s syndrome has a very strong association with type 1 RTA and should be considered in the differential diagnosis even in the absence of sicca syndrome.
In type 2 RTA, the proximal tubule has a reduced capacity to reabsorb HCO3–. As the kidneys lose HCO3– and plasma HCO3– concentration falls, the filtered load of HCO3– decreases reaching a level that can be reabsorbed and a new steady state of serum HCO3– concentration is reached. Because of this ability of the kidney to decrease HCO3– excretion as well as compensatory increase in H+ secretion by the distal tubules, despite the aminoaciduria, urinary pH is <5.5. Reabsorption of Na+ (linked to reclaiming of HCO3–) in the proximal tubules helps to cotransport amino acid, glucose, uric acid, and phosphate. Type 2 RTA can occur as an isolated defect in HCO3– reabsorption or as a generalized defect in proximal tubular reabsorption with aminoaciduria, phosphaturia, glycosuria, and uric aciduria when the condition is termed Fanconi syndrome. The patient with Fanconi syndrome would present with normal anion gap metabolic acidosis with hypokalemia, urine pH <5.5, glycosuria, proteinuria, and low plasma phosphate and uric acid.
Although type 2 RTA may be familial, one must always suspect myeloma where the filtered immunoglobulin light chains by causing PCT cellular toxicity can lead to this condition. Other causes include Wilson’s disease, cystinosis, Lowe syndrome, outdated tetracycline, carbonic anhydrase inhibitors such as acetazolamide and topiramate, and lead or mercury poisoning (Table 2).
The metabolic acidosis in both types 1 and 2 RTA causes bones to release calcium salts to function as a buffer. This leads to progressive osteopenia and osteomalacia and also increased urinary calcium excretion. Under normal circumstances urinary citrate binds with calcium and makes it soluble. However, citrate is an effective alkaline buffer and hence in type 1 RTA, the proximal tubules tend to reabsorb increased amounts of citrate in an effort to combat the acidosis. The resultant deficiency of citrate in urine in type 1 RTA tends to make the urinary calcium insoluble. In addition, the alkaline urine markedly diminishes the urinary calcium solubility. The end result is increased incidence of nephrocalcinosis and nephrolithiasis in type 1 RTA.
As both types 1 or 2 RTA present with normal anion gap metabolic acidosis and hypokalemia, the only definitive way to distinguish between the two conditions is the urine pH (Table 3). Type 1 RTA is characteristically associated with urinary pH >5.5. In contrast, the urinary pH is usually <5.5 in type 2 RTA except when the patient is being treated with massive doses of alkali. Hypophosphatemia, hypouricemia, and glycosuria in the absence of raised blood glucose point toward Fanconi syndrome.
NORMAL ANION GAP (HYPERCHLOREMIC) METABOLIC ACIDOSIS IN DIARRHEA
Chronic diarrheal states may present with normal anion gap metabolic acidosis and hypokalemia and hence simulate RTA. The mechanism is as follows:
- Metabolic acidosis: Due to fecal loss of bicarbonate-rich pancreatic secretions
- Hypokalemia: Hypovolemia can lead to activation of renin–angiotensin–aldosterone system (RAAS) with the resultant tubular loss of K+ under the influence of aldosterone.
The urine anion gap (UAG) calculation helps to differentiate between RTA type 1 and extrarenal causes of normal anion gap acidosis such as diarrheal states, being positive in the former and negative in the latter.
UAG (in mEq/L or mmol/L) = Urine (Na+ + K+ – Cl–)
The UAG is positive (between 20 and 90) in healthy individuals because the dietary and hence urinary Na+ and K+ is normally more than Cl–. With metabolic acidosis from any cause, the kidney will respond by excreting a heavy load of H+ which will also need increased ammonia to buffer the H+. Since ammonia in the urine exists as NH4Cl, this leads to increased urinary Cl– with the result that the UAG becomes negative. In type 1 RTA since the defect is in the secretion of H+ and therefore there is no need for extra ammonia and hence urinary NH4Cl remains low and UAG remains positive.
Management
Treatment of both types 1 and 2 RTA consists of potassium replacement and alkali therapy. One should be careful when administering NaHCO3 because the resultant Na+ loss in urine may increase K+ secretion and thus exacerbate hypokalemia. In this setting, it is advisable to replace K+ before the acidosis is corrected. Potassium citrate is often useful in these patients.
As explained above, type 1 RTA patients have low urinary citrate which makes them more predisposed to recurrent urinary stones. Potassium citrate can be useful in this patient group too.
Type 2 RTA often needs much bigger doses of alkali because of the tendency of the kidneys to excrete more HCO3– as the serum HCO3– concentration increases.
TYPE 4 RTA (HYPERKALEMIC DISTAL TUBULAR ACIDOSIS)
Type 4 RTA is the most common among the RTAs and patients present with normal anion gap metabolic acidosis and hyperkalemia.
Under normal circumstances, secretion of H+ and K+ in the collecting duct is dependent on the intraluminal negativity generated as a consequence of aldosterone-induced reabsorption of the Na+. With hypoaldosteronism or tubular unresponsiveness to aldosterone, less Na+ is reabsorbed and hence less H+ and K+ are secreted into the tubules.
Hyperkalemia in itself causes decreased tubular secretion of H+ by decreasing renal ammonia generation; ammonia is one of the chief buffers for H+ in the urine. Ammonia is derived from breakdown of glutamine by phosphate-dependent glutaminase (PDG) in the PCT cells. Hyperkalemia decreases PDG activity and therefore decreases ammonia production which in turn interferes with the tubular capacity to secrete H+.
The etiology of type 4 RTA can be divided in three groups based on the pathophysiology:
1. Aldosterone deficiency: Hyporeninemic hypoaldosteronism is common in diabetes mellitus. Other causes of hypoaldosteronism include nonsteroidal anti-inflammatory drug (NSAID) use, calcineurin inhibitors (cyclosporine and tacrolimus), angiotensin-converting enzyme (ACE) inhibitor or angiotensin receptor blocker (ARB) groups of drugs, and heparin or low molecular heparin.
2. Tubular unresponsiveness or resistance to aldosterone: This is seen with:
a. Potassium-sparing diuretics by either antagonizing the action of aldosterone by competing for the aldosterone receptor (spironolactone and eplerenone) or closing the sodium channels in the luminal membrane (amiloride and triamterene).
b. Antibiotics: Trimethoprim and pentamidine can cause type 4 RTA by closing the epithelial sodium channels in the collecting tubule with the resultant inability to excrete K+ and H+.
3. Less commonly acquired or inherited defect in the sodium transport mechanisms in the principal cells: Some patients with severe long-standing volume contraction due, for example, to long-term laxative abuse may develop a pure voltage-dependent RTA. Persistent hypovolemia causes excess Na+ reabsorption by the early tubules leaving less Na+ available in the urine as it reaches the collecting duct; secretion of K+ and H+ in this part of the tubule is linked to reabsorption of Na+. Sickle cell disease and lithium cause type 4 RTA by this mechanism.
MANAGEMENT
In patients who are not hypertensive or volume overloaded, administration of a synthetic mineralocorticoid such as fludrocortisone may be effective. In patients with hypertension or fluid overload, a thiazide or loop diuretic may help by increasing distal delivery of Na+ and consequently increasing the secretion of H+ and K+.
CONCLUSION
Renal tubular acidosis refers to a group of disorders characterized by normal anion gap (hyperchloremic) metabolic acidosis with either hypokalemia or hyperkalemia and a relatively well preserved GFR. Proximal RTA (type 2 RTA) is caused by reduced ability to reabsorb HCO3– in the proximal tubules while distal RTA (type 1 RTA) is caused by defect in distal H+ excretion. Type 1 RTA is characteristically associated with urinary pH >5.5 while the urine pH is <5.5 in type 2 RTA. Sjögren’s syndrome has a strong association with type 1 RTA. One must always consider myeloma in the differential in patients presenting with type 2 RTA.
While both the conditions are treated with alkali and potassium replacement, type 2 RTA often needs larger doses of alkali which sometimes can cause worsening hypokalemia. In these patients and the type 1 RTA patients who are recurrent renal calcium stone formers, one can use potassium citrate for alkali replacement.
Type 4 RTA is mostly caused due to either aldosterone deficiency or tubular resistance to the action of aldosterone. It is quite common in those with diabetes and also associated with drugs which interfere with the RAAS pathway, i.e., ACE inhibitor or ARB as well as aldosterone antagonists such as spironolactone. While synthetic mineralocorticoids such as fludrocortisone are effective, in patients with hypertension or fluid overload, thiazide or loop diuretic may be helpful.
SUGGESTED READINGS
1. Weiner ID, Verlander JW. Renal ammonia metabolism and transport. Compr Physiol. 2013;3(1):201-20.
2. Sarah S, Lijo G, Sukanya E, Rajasekaran D. Renal tubular acidosis due to Sjogren’s syndrome presenting as hypokalemic quadriparesis: A report of two cases. Indian J Nephrol. 2015;25(6):386-7.
3. Rodríguez Soriano J. Renal tubular acidosis: the clinical entity. J Am Soc Nephrol. 2002;13(8):2160-70.
4. Santos F, Ordóñez FA, Claramunt-Taberner D, Gil-Peña H. Clinical and laboratory approaches in the diagnosis of renal tubular acidosis. Pediatr Nephrol. 2015;30(12):2099-107.
5. Palmer BF, Kelepouris E, Clegg DJ. Renal Tubular Acidosis and Management Strategies: A Narrative Review. Adv Ther. 2021;38(2):949-68.
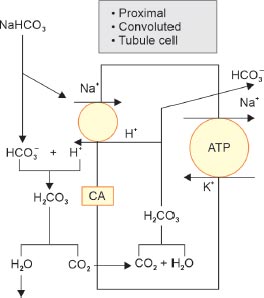
FIG. 1: Action of carbonic anhydrase (CA) in reclaiming HCO3– in the proximal convoluted tubule (PCT).
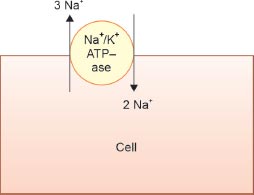
FIG. 2: The Na+-K+-ATPase extrudes 3 Na+ and pulls in 2 K+ into cells leading to the cells being low in Na+ and high in K+ and also electronegative (lose three positively charged Na+ but gain only 2 K+).
| TABLE 1: Causes of type 1 RTA. | |
Autoimmune disorders |
• Sjögren’s syndrome (most common) • SLE • Autoimmune hepatitis/primary biliary cirrhosis • Rheumatoid arthritis |
Drugs |
Amphotericin, lithium, ibuprofen, and ifosfamide (can also cause type 2 RTA) |
Hypercalciuric conditions |
Hyperparathyroidism, vitamin D intoxication, and sarcoidosis |
Miscellaneous causes |
Renal transplantation, obstructive uropathy, and medullary sponge kidney |
| (RTA: renal tubular acidosis; SLE: systemic lupus erythematosus) | |
| TABLE 2: Causes of type 2 renal tubular acidosis (RTA). | |
M protein disorders |
• Multiple myeloma • Light chain disease • Amyloidosis |
Drugs |
• Tenofovir and related antiretroviral drugs • Carbonic anhydrase inhibitors—acetazolamide and topiramate • Outdated tetracycline • Ifosfamide (can also cause type 1 RTA) • Cisplatin/Oxaliplatin |
Heavy metals |
• Lead • Cadmium • Copper • Mercury |
Familial |
• Wilson disease • Lowe syndrome • Cystinosis • Tyrosinemia |
Miscellaneous causes |
• Paroxysmal nocturnal hemoglobinuria • Severe vitamin D deficiency or resistance |
| TABLE 3: Differentiating features between types 1 and 2 renal tubular acidosis (RTA). | |
Type 1 RTA (distal RTA) |
Type 2 RTA (proximal RTA) |
Inability to secrete H+ |
Inability to reabsorb HCO3– |
Urine pH >5.5 (low H+ in urine) |
Urine pH <5.5 despite bicarbonaturia (compensatory increase in distal tubular H+ secretion) |
Nephrolithiasis and nephrocalcinosis |
No renal stones |
No Fanconi syndrome |
May have Fanconi syndrome: glycosuria, phosphaturia, uric aciduria, and aminoaciduria |
Treat with alkali and K+ replacement |
Same treatment but often need bigger doses of alkali |